RASopathy Syndromes
Scroll down to find information about and resources for the following germline mutation syndromes:
- CFC syndrome (CFCS)
- Costello syndrome (CS)
- Neurofibromatosis type 1 (NF1)
- Legius syndrome
- Noonan Syndromes
- Noonan syndrome (NS)
- Noonan syndrome with multiple lentigines (NSML)
- Noonan syndrome-like disorder with loose anagen hair (NSLH)
- Noonan syndrome-like disorder with or without juvenile myelomonocytic leukemia (NSLL)
Also see Other RASopathy Syndromes: CM-AVM and SYNGAP1-ID, and HINCONS
CFC syndrome

1:810,000 (Abe, Aoki et al., 2012); 1:150,000 (unpublished, UK)
Genes: BRAF, KRAS, MAP2K1 (MEK1), MAP2K2 (MEK2)
Cardiofaciocutaneous syndrome (CFCS) is characterized by cardiac abnormalities (pulmonic stenosis and other valve dysplasias, septal defects, hypertrophic cardiomyopathy, rhythm disturbances), distinctive craniofacial appearance, and cutaneous abnormalities (including xerosis, hyperkeratosis, ichthyosis, keratosis pilaris, ulerythema ophryogenes, eczema, pigmented moles, hemangiomas, and palmoplantar hyperkeratosis). The hair is typically sparse, curly, fine or thick, woolly or brittle; eyelashes and eyebrows may be absent or sparse. Nails may be dystrophic or fast growing. Some form of neurologic and/or cognitive delay (ranging from mild to severe) is seen in all affected individuals. Neoplasia, mostly acute lymphoblastic leukemia (ALL), has been reported in some individuals. [Rauen KA, GeneReviews for CFC syndrome, 2016]
External Website Links:
- CFC International
- CFC Parent Guide by Pilar Magoulas, MS, CGC for CFC International
- 2023 CFC GeneReveiw
- Genetic Home Reference for CFC syndrome
- CFC Syndrome Clinical Features, Diagnosis and Medical Management, Pediatrics, 2014
Costello syndrome (CS)

1:1.29 million (Abe, Aoki et al., 2012); 1:300,000 (Gripp et al., 2019)
Gene: HRAS
Costello syndrome affects multiple organ systems. Its typical presentation is characterized by diffuse hypotonia and severe feeding difficulties in infancy; short stature; developmental delay or intellectual disability; characteristic facial features; curly or sparse, fine hair; loose, soft skin with deep palmar and plantar creases; papillomata of the face and perianal region; joint laxity with ulnar deviation of the wrists and fingers; tight Achilles tendons; and cardiac involvement (hypertrophic cardiomyopathy [HCM], congenital heart defect, and arrhythmia). Postnatal cerebellar overgrowth can result in Chiari I malformation with associated hydrocephalus or syringomyelia. An approximately 15% lifetime risk for malignant tumors includes rhabdomyosarcoma and neuroblastoma in young children and transitional cell carcinoma of the bladder in adolescents and young adults. Females and males are equally affected. [Gripp KW and Rauen KA, GeneReviews for CS, 2019]
External Costello syndrome family support website links:
Publications for families:
- CS Booklet by Parents for Parents, 11 pages
- 父母为父母提供的 Costello 综合症小册子
- Livret costello (en français), 60 pages
- CS Guidelines for Clinical Diagnosis trifold
- 2022 Multidisciplinary Management of Costello Syndrome by Chiara Leoni. Table 2 shows timing for clinical and instrumental follow-up, according to literature and the authors’ experience
- Síndrome de Costello ~ Guía de diagnosis clinica tríptico
External website links about Costello syndrome:
Neurofibromatosis Type 1 (NF1)

1:2,000 to 1:5,000
Gene: NF1
Neurofibromatosis 1 (NF1) is characterized by multiple café au lait spots, axillary and inguinal freckling, multiple cutaneous neurofibromas, and iris Lisch nodules. About half of people with NF1 have plexiform neurofibromas, but most are internal and not suspected clinically. Learning disabilities are present in at least 50% of individuals with NF1. Less common but potentially more serious manifestations include optic nerve and other central nervous system gliomas, malignant peripheral nerve sheath tumors, scoliosis, tibial dysplasia, and vasculopathy. [Friedman JM, GeneReviews for Neurofibromatosis 1, 2019]
External Website Links:
- Children’s Tumor Foundation
- NF Network
- GeneReviews for NF1
- Genetic Home Reference for NF1
- Whiteboard video explaining NF1
Legius syndrome (formerly NF1-like syndrome; NFLS)
Gene: SPRED1
Legius syndrome is characterized by multiple café au lait macules without neurofibromas or other tumor manifestations of neurofibromatosis type 1 (NF1). Additional clinical manifestations reported commonly include intertriginous freckling, lipomas, macrocephaly, and learning disabilities / ADHD / developmental delays. Current knowledge of the natural history of Legius syndrome is based on the clinical manifestations of fewer than 200 individuals with a molecularly confirmed diagnosis; better delineation of the clinical manifestations and natural history of Legius syndrome will likely occur as more affected individuals are identified. [Stevenson D, Viskochil D, Mao R, GeneReviews for Legius syndrome, 2015]
External Website Links:
Hereditary Gingival Neurofibromatosis Type 1
Gingival fibromatosis is a rare overgrowth condition characterized by a benign, slowly progressive, nonhemorrhagic, fibrous enlargement of maxillary and mandibular keratinized gingiva [summary by Hart et al., 2002].
External Website Link:
Noonan Syndromes
Noonan syndrome (NS)
 1:1,000 to 1:2,500
1:1,000 to 1:2,500
Genes: BRAF, CBL, KRAS, LZTR1, MAP2K1 (MEK1), MRAS, NRAS, PTPN11, RAF1, RIT1, RRAS, SOS1, SOS2
Noonan syndrome (NS) is characterized by characteristic facies, short stature, congenital heart defect, and developmental delay of variable degree. Other findings can include broad or webbed neck, unusual chest shape with superior pectus carinatum and inferior pectus excavatum, cryptorchidism, varied coagulation defects, lymphatic dysplasias, and ocular abnormalities. Although birth length is usually normal, final adult height approaches the lower limit of normal. Congenital heart disease occurs in 50%-80% of individuals. Pulmonary valve stenosis, often with dysplasia, is the most common heart defect and is found in 20%-50% of individuals. Hypertrophic cardiomyopathy, found in 20%-30% of individuals, may be present at birth or develop in infancy or childhood. Other structural defects include atrial and ventricular septal defects, branch pulmonary artery stenosis, and tetralogy of Fallot. Up to one fourth of affected individuals have mild intellectual disability, and language impairments in general are more common in NS than in the general population. [Allanson JE and Roberts AE, GeneReviews for Noonan syndrome, 2019]
External Website Links:
- Noonan Syndrome Foundation (NSF) – a USA-registered family support group for people with Noonan syndrome
- Noonan Syndrome Association, a UK-registered Charity supporting people with Noonan syndrome
- Association Noonan – a French support organization with a scientific advisory board
- Noonan Syndrome Awareness Association – Australian group support and information
- Noonan syndrome list of internet resources curated by Marla Wessland
- GeneReviews for Noonan syndrome
- Genetics Home Reference for Noonan syndrome
- Management of Noonan Syndrome (2011) by DYSCERNE – Noonan Syndrome Management Development Group
- A guide for teachers: Attention and executive function concerns in children with Noonan syndrome by the MIND group at University of Minnesota
- NS Growth Charts: for Boys and for Girls
Noonan syndrome with multiple lentigines (NSML) (formerly LEOPARD syndrome)
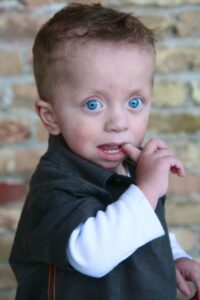
Genes: PTPN11, RAF1, BRAF, MAP2K1 (MEK1)
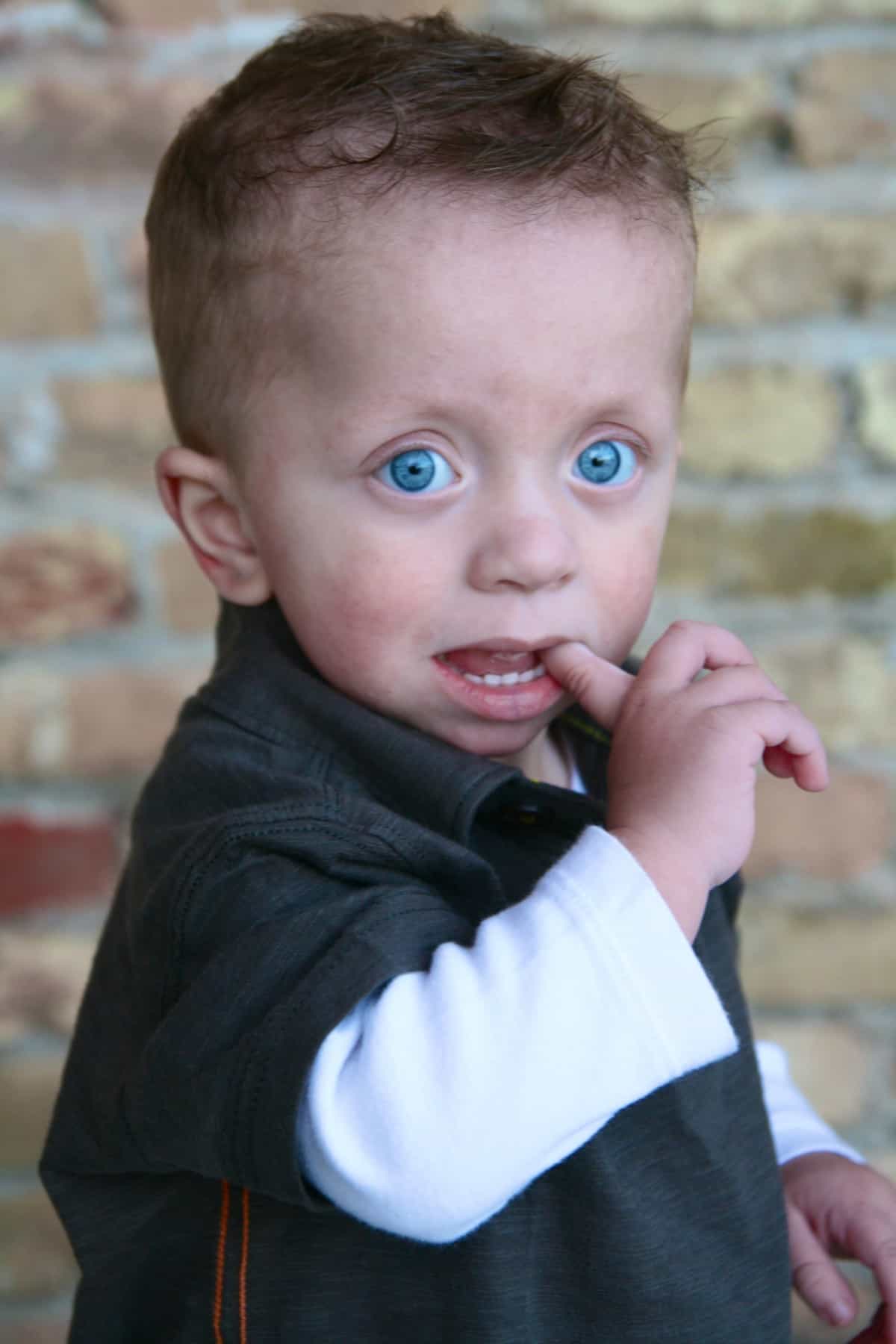
Noonan syndrome with multiple lentigines (NSML) is a condition in which the cardinal features consist of lentigines, hypertrophic cardiomyopathy, short stature, pectus deformity, and dysmorphic facial features, including widely spaced eyes and ptosis. Multiple lentigines present as dispersed flat, black-brown macules, mostly on the face, neck and upper part of the trunk with sparing of the mucosa. In general, lentigines do not appear until age four to five years but then increase to the thousands by puberty. Some individuals with NSML do not exhibit lentigines. Approximately 85% of affected individuals have heart defects, including hypertrophic cardiomyopathy (HCM) (typically appearing during infancy and sometimes progressive) and pulmonary valve stenosis. Postnatal growth retardation resulting in short stature occurs in fewer than 50% of affected persons, although most affected individuals have a height that is less than the 25th percentile for age. Sensorineural hearing deficits, present in approximately 20%, are poorly characterized. Intellectual disability, typically mild, is observed in approximately 30% of persons with NSML. [Gelb BD and Tartaglia M, GeneReviews for Noonan Syndrome with Multiple Lentigines, 2015]
External Website Links:
- GeneReviews for NSML syndrome
- Genetic Home Reference for Multiple lentigines syndrome
- NSML Facebook group
Noonan syndrome-like disorder with loose anagen hair (NSLH or NSLAH)
 Genes: SHOC2 (for NSLH1), PPP1CB (for NSLH2)
Genes: SHOC2 (for NSLH1), PPP1CB (for NSLH2)
Paraphrased from the OMIM entry for NSLH1:
Noonan syndrome-like disorder with loose anagen hair is characterized by facial features similar to those observed in Noonan syndrome (163950), including hypertelorism, ptosis, downslanting palpebral fissures, low-set posteriorly angulated ears, and overfolded pinnae. In addition, patients display short stature, frequently with growth hormone (GH; see 139250) deficiency; cognitive deficits; relative macrocephaly; small posterior fossa resulting in Chiari I malformation; hypernasal voice; cardiac defects, especially dysplasia of the mitral valve and septal defects; and ectodermal abnormalities, in which the most characteristic feature is the hair anomaly, including easily pluckable, sparse, thin, slow-growing hair (summary by Bertola et al., 2017).
Mazzanti et al. (2003) suggested that the disorder in these children is distinct from Noonan syndrome.
External website links:
- OMIM entry for NSLH1
- Genetic and Rare Disease (GARD) Information Center NSLH
- ORPHANET listing for NS/LAH
- OMIM entry for NSLH2
Noonan syndrome-like disorder with or without juvenile myelomonocytic leukemia (NSLL)
 Gene: CBL Other Names: Cbl syndrome
Gene: CBL Other Names: Cbl syndrome
Noonan syndrome-like disorder is a developmental disorder resembling Noonan syndrome (NS1; 163950) and characterized by facial dysmorphism, a wide spectrum of cardiac disease, reduced growth, variable cognitive deficits, and ectodermal and musculoskeletal anomalies. There is extensive phenotypic heterogeneity and variable expressivity (summary by Martinelli et al., 2010). Patients with heterozygous germline CBL mutations have an increased risk for certain malignancies, particularly juvenile myelomonocytic leukemia (JMML; 607785), as also seen in patients with Noonan syndrome (summary by Niemeyer et al., 2010). [OMIM]
External website links:
Other RASopathy Syndromes
Capillary malformation-arteriovenous malformation syndrome (CM-AVM)
Gene: RASA1
Capillary malformation-arteriovenous malformation (CM-AVM) syndrome is characterized by the presence of multiple small (1-2 cm in diameter) capillary malformations mostly localized on the face and limbs. Some affected individuals also have associated arteriovenous malformations (AVMs) and/or arteriovenous fistulas (AFVs), fast-flow vascular anomalies that typically arise in the skin, muscle, bone, spine, and brain; life-threatening complications of these lesions can include bleeding, congestive heart failure, and/or neurologic consequences. Symptoms from intracranial AVMs/AVFs appear to occur early in life. Several individuals have Parkes Weber syndrome (multiple micro-AVFs associated with a cutaneous capillary stain and excessive soft-tissue and skeletal growth of an affected limb). [Bayrak-Toydemir P and Stevenson D, GeneReviews for CM-AVM, 2019]
- GeneReviews for CM-AVM syndrome
- OMIM entry for CMAVM1
- Genetic Home Reference for CM-AVM syndrome
- ARUP Laboratories; University of Utah RASA1 Home
SYNGAP1-related intellectual disability (SYNGAP1-ID)
Gene: SYNGAP1
SYNGAP1-related intellectual disability (SYNGAP1-ID) is characterized by developmental delay (DD) or intellectual disability (ID) (100% of affected individuals), generalized epilepsy (~84%), and autism spectrum disorder (ASD) and other behavioral abnormalities (≤50%). To date more than 50 individuals with SYNGAP1-ID have been reported. In the majority DD/ID was moderate to severe; in some it was mild. The epilepsy is generalized; a subset of individuals with epilepsy have myoclonic astatic epilepsy (Doose syndrome) or epilepsy with myoclonic absences. Behavioral abnormalities can include stereotypic behaviors (e.g., hand flapping, obsessions with certain objects) as well as poor social development. Feeding difficulties can be significant in some. [Holder JL, Hamdan FF, and Michaud JL, GeneReviews for SYNGAP1-ID, 2019]
Hiatt-Neu-Cooper neurodevelopmental syndrome (HINCONS)
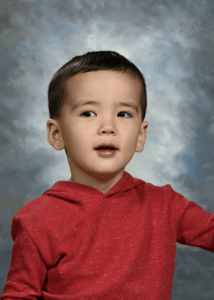
Gene: RALA
From the OMIM entry for HINCONS
Hiatt-Neu-Cooper neurodevelopmental syndrome (HINCONS) is characterized by global developmental delay with delayed walking or inability to walk and impaired intellectual development with poor or absent speech. Affected individuals have axial hypotonia and dysmorphic facies. Additional more variable features may include seizures, autistic or behavioral abnormalities, and brain abnormalities, such as dysplastic corpus callosum or polymicrogyria. [Hiatt et al., 2018]
Parents of Arlo are seeking other families affected by RALA to start a community to build awareness in hopes of inspiring researcher interest.

